Diseño de ensayo cruzado: cómo se estructuran los estudios de bioequivalencia
 mar, 20 2026
mar, 20 2026
Los estudios de bioequivalencia son fundamentales para aprobar medicamentos genéricos. Pero no todos los diseños funcionan igual. El diseño de ensayo cruzado es el método más usado en el mundo, y por una buena razón: es más eficiente, preciso y económico que otros enfoques. Si quieres entender por qué los reguladores lo prefieren, y cómo se organiza realmente un estudio de este tipo, este es el mapa detallado que necesitas.
¿Qué es un ensayo cruzado y por qué se usa en bioequivalencia?
Imagina que en lugar de comparar dos grupos de personas -uno toma el medicamento genérico, otro el original-, cada persona prueba ambos medicamentos, uno tras otro. Eso es un ensayo cruzado. Cada participante actúa como su propio control. Esto elimina las diferencias individuales que arruinan otros estudios: edad, peso, metabolismo, genética. En un estudio paralelo, si un grupo tiene más personas con hígado lento, los resultados se distorsionan. En un cruzado, eso no importa. Todos experimentan ambos tratamientos, y solo se mide la diferencia entre ellos.
Este diseño no es nuevo. Se usó por primera vez en los años 50, pero se volvió estándar en bioequivalencia después de las guías de la FDA en 1992 y la EMA en 2010. Hoy, el 89% de los estudios de bioequivalencia aprobados por la FDA usan este modelo. ¿Por qué? Porque reduce el número de participantes necesarios hasta en un 83%. Si un estudio paralelo necesita 72 personas, el cruzado puede hacerlo con 24. Menos voluntarios, menos costos, menos tiempo. Y la precisión sube.
El diseño básico: 2 períodos, 2 secuencias (2×2)
El modelo más simple y más común es el 2×2. Cada participante recibe dos tratamientos: el producto de prueba (el genérico) y el producto de referencia (el original). Pero no todos lo prueban en el mismo orden. Se dividen en dos grupos:
- Grupo AB: primero el genérico, luego el original.
- Grupo BA: primero el original, luego el genérico.
Entre un tratamiento y otro, hay un periodo de lavado. Este es crítico. Debe durar al menos cinco veces la vida media del fármaco. Si el medicamento se elimina del cuerpo en 8 horas, el lavado debe ser de al menos 40 horas. Esto asegura que el fármaco del primer periodo haya desaparecido completamente antes de empezar el segundo. Si no, los resultados se contaminan: lo que se mide en el segundo periodo no es solo el nuevo medicamento, sino una mezcla de ambos. Eso hace que el estudio falle.
Después de cada periodo, se toman muestras de sangre varias veces para medir la concentración del fármaco. Se calculan dos parámetros clave: el área bajo la curva (AUC), que indica cuánto fármaco se absorbió en total, y la concentración máxima (Cmax), que muestra qué tan rápido se absorbió. Para que el genérico sea bioequivalente, la relación de las medias geométricas (genérico/original) debe estar entre el 80% y el 125% para ambos parámetros. Si cae fuera, el estudio no pasa.
¿Y si el medicamento es muy variable?
No todos los fármacos son iguales. Algunos, como la warfarina o el fenitoína, tienen una variabilidad interna muy alta. Eso significa que incluso en la misma persona, las concentraciones de sangre pueden cambiar mucho de una dosis a otra. En estos casos, el diseño 2×2 no es suficiente. La variabilidad interna supera el 30% (CV >30%), y el intervalo de confianza del 80-125% ya no es confiable.
Entonces se usan diseños replicados. Hay dos tipos:
- Parcialmente replicado (TRR/RTR): El participante recibe el genérico dos veces y el original una vez (o viceversa). Por ejemplo: TRR (genérico, original, genérico) y RTR (original, genérico, original). Esto permite estimar la variabilidad interna del genérico y del original por separado.
- Completamente replicado (TRTR/RTRT): Cada medicamento se administra dos veces. Esto da una estimación aún más precisa.
Estos diseños permiten usar un enfoque llamado RSABE (bioequivalencia promedio escalada por referencia). En vez de usar siempre el 80-125%, se ajusta el rango según la variabilidad real del fármaco. Para algunos medicamentos muy variables, se puede aceptar un rango de 75-133%. Sin estos diseños replicados, los estudios fallarían constantemente, incluso si el genérico era igual. Hoy, el 47% de los medicamentos muy variables aprobados por la FDA usan este método. En 2024, la EMA lo hará obligatorio para todos.
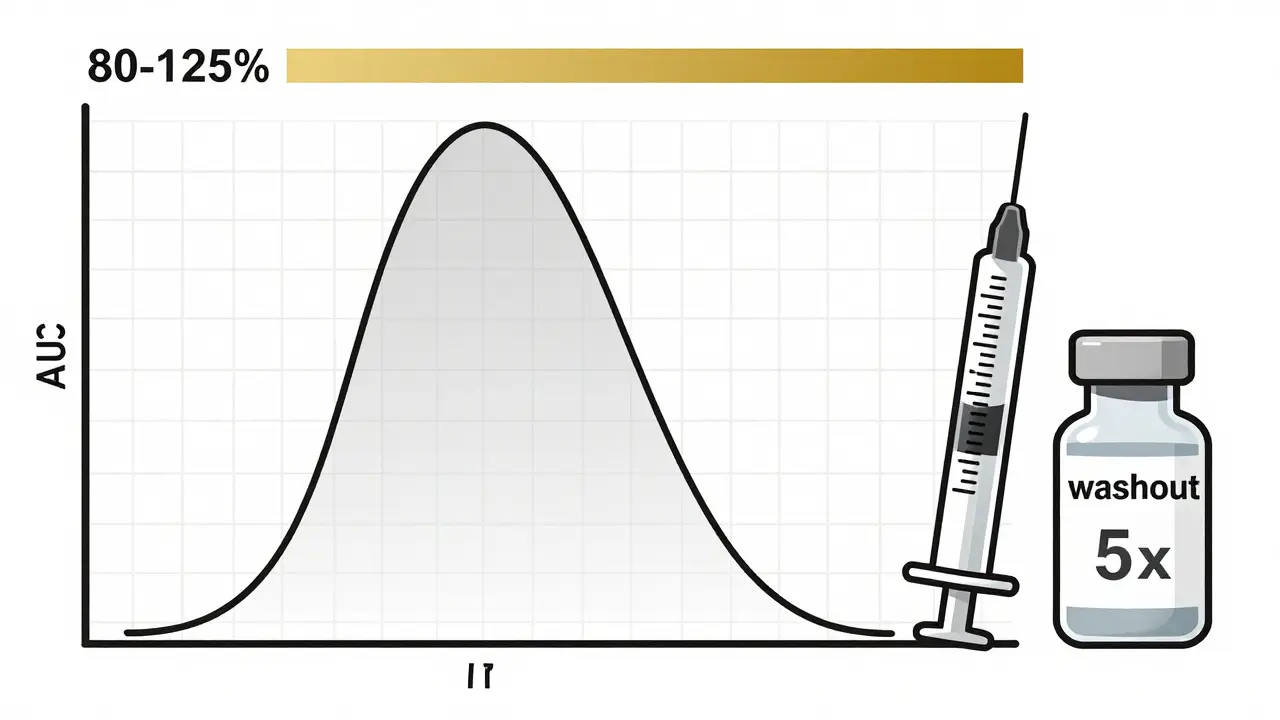
¿Por qué no usar siempre el diseño replicado?
Porque tiene un costo. Un estudio replicado de 4 períodos lleva más tiempo, más visitas, más muestras de sangre, más recursos. Puede costar hasta un 40% más que un 2×2 simple. Además, la tasa de abandono sube: más visitas = más personas que se retiran. Por eso, no se usa para todo. Solo cuando la variabilidad lo exige.
Un estudio de 2021 en ResearchGate contó cómo un equipo falló porque usó un lavado de solo 3 días para un fármaco con vida media de 18 horas. La concentración residual afectó el segundo periodo. Tuvo que repetirse con un diseño replicado, a un costo extra de $195,000. Eso no es raro. El 15% de las negaciones de estudios por la FDA en 2018 se debieron a lavados inadecuados.
Errores comunes que arruinan los estudios
El diseño cruzado es poderoso, pero frágil si se hace mal. Aquí van los errores más frecuentes:
- Lavado insuficiente: El más común. No se calcula bien la vida media o se ignora la variabilidad individual.
- Secuencias desbalanceadas: Si hay más personas en AB que en BA, se introduce un sesgo de periodo.
- Errores estadísticos: Usar modelos simples en lugar de modelos mixtos (como PROC MIXED en SAS). No ajustar por periodo o secuencia.
- Perder datos: Si un participante se retira después del primer periodo, se pierde la ventaja del diseño cruzado. No se puede imputar bien sin sesgar.
- Ignorar la variabilidad: Aplicar el 80-125% a un medicamento con CV del 45% sin usar RSABE.
Las empresas que usan software como Phoenix WinNonlin tienen ventaja: tienen plantillas validadas. Pero quienes usan R o Python deben tener estadísticos con experiencia. Un error en el modelo puede hacer que un buen genérico parezca malo.
El futuro: ¿Qué cambia en 2026?
La tendencia es clara: más diseños replicados. La FDA ya permite diseños de 3 períodos para medicamentos de índice terapéutico estrecho. La EMA lo hará obligatorio en 2024. Y los estudios adaptativos están ganando terreno: ahora el 23% de los estudios aprobados en la FDA en 2022 incluyeron reestimación de tamaño de muestra en una segunda etapa. Esto significa que si al final del primer periodo la variabilidad es mayor de lo esperado, se añaden más participantes sin arruinar el estudio.
¿Desaparecerá el diseño 2×2? No. Sigue siendo el estándar para el 68% de los medicamentos. Pero el futuro pertenece a los diseños que pueden manejar la complejidad. Los medicamentos más nuevos -como los biosimilares, los antirretrovirales o los fármacos para enfermedades raras- son cada vez más variables. Necesitan más datos, más precisión, más flexibilidad.
Y aunque la tecnología digital promete monitoreo continuo (sensores que miden fármacos en sangre sin pinchazos), todavía no reemplaza los ensayos cruzados. Por ahora, el diseño cruzado sigue siendo el oro estándar. Y lo será, al menos, hasta 2035.
¿Cuándo NO se usa el diseño cruzado?
No es universal. Hay casos donde es imposible:
- Fármacos con vida media muy larga (más de 2 semanas). Un lavado de 14 días es inviable para un voluntario.
- Enfermedades crónicas donde el tratamiento debe ser continuo (por ejemplo, antidepresivos o antiepilépticos).
- Medicamentos con efectos irreversibles o daños permanentes.
- Estudios en niños o ancianos con alta tasa de abandono.
En esos casos, se recurre al diseño paralelo. Pero es la excepción. Y siempre requiere más participantes, más dinero, más tiempo.
¿Cuál es el tamaño típico de muestra en un estudio de bioequivalencia con diseño cruzado?
En un diseño 2×2 estándar, se necesitan entre 12 y 48 participantes, dependiendo de la variabilidad del fármaco. Si la variabilidad interna (CV) es baja (10-15%), 12-24 sujetos pueden ser suficientes. Si es media (15-25%), se usan 24-36. Para fármacos con CV de 30-35%, se requieren 36-48. En diseños replicados, los tamaños aumentan a 24-72 participantes, pero permiten estudiar medicamentos muy variables que de otro modo serían inviables.
¿Qué pasa si un participante se retira después del primer periodo?
Si un participante se retira después del primer periodo, sus datos se excluyen del análisis. Esto es porque el diseño cruzado depende de comparar los mismos individuos entre tratamientos. Sin ambos periodos, no se puede hacer la comparación dentro del sujeto. Aunque esto reduce el tamaño de la muestra, no invalida el estudio si el número restante sigue siendo estadísticamente poderoso. Lo que sí invalida el estudio es intentar imputar datos faltantes con modelos estadísticos que asumen una correlación que no existe.
¿Por qué se usa la media geométrica y no la media aritmética en los estudios de bioequivalencia?
Porque las concentraciones de fármacos en sangre se distribuyen de forma log-normal, no normal. Es decir, los valores no están repartidos de forma simétrica, sino con una cola larga hacia arriba. La media aritmética se distorsiona con valores extremos. La media geométrica, al trabajar en escala logarítmica, es más robusta y representa mejor la tendencia central de los datos farmacocinéticos. Por eso, los reguladores exigen comparar las medias geométricas de AUC y Cmax.
¿Qué es el periodo de lavado y cómo se determina?
El periodo de lavado es el tiempo entre dos tratamientos en un ensayo cruzado, diseñado para eliminar completamente el fármaco del primer periodo. Se calcula multiplicando la vida media del fármaco por al menos cinco. Por ejemplo, si la vida media es 6 horas, el lavado debe ser de 30 horas. Esto se basa en que tras cinco vidas medias, más del 97% del fármaco se ha eliminado. Se valida con estudios piloto o datos de literatura, y siempre se mide la concentración residual antes del segundo periodo para confirmar que está por debajo del límite de cuantificación.
¿Qué porcentaje de estudios de bioequivalencia usan diseños replicados hoy?
En 2023, aproximadamente el 25% de todos los estudios de bioequivalencia aprobados por la FDA usaban diseños replicados (parciales o completos). De ellos, el 22% eran diseños parciales (TRR/RTR) y el 10% eran completos (TRTR/RTRT). Esta cifra ha crecido del 12% en 2015 al 47% en 2022 para medicamentos muy variables. Se espera que en 2026, el 40% de todos los estudios utilicen algún tipo de diseño replicado, especialmente con el aumento de medicamentos complejos y biosimilares.

Sheila Ruiz
marzo 20, 2026 AT 16:52Patricia C Perez
marzo 20, 2026 AT 16:53Hernán Rivas
marzo 22, 2026 AT 04:33Valentina Capra
marzo 22, 2026 AT 22:14África Barragán Quesada
marzo 23, 2026 AT 07:52Javier Martínez Misol
marzo 24, 2026 AT 03:15Yessenia Quiros Montoya
marzo 25, 2026 AT 18:42Marvin Ameth Barrios Becerra
marzo 27, 2026 AT 05:25